Guillain-Barré syndrome (GBS) is a post-infectious and immune-mediated polyneuropathy and is a clinically heterogeneous condition divided into demyelinating and axonal categories.1,2 Approximately 60% of patients with GBS are positive for anti-ganglioside antibodies.3 Anti-GM1 and -GD1a antibodies are markers of an immune response primarily affecting the motor axons of the peripheral nervous system and closely related to the acute motor axonal neuropathy (AMAN) variant of GBS.2,4 Considering the possible pathogenesis of AMAN subsequent to Campylobacter jejuni infection, the binding of antibodies to GM1 or GD1a at nodes of Ranvier in peripheral motor nerves induces local complement activation, resulting in the formation of membrane-attack complex, axonal degeneration, and loss of myelin. This axonal injury is thought to be associated with poor outcomes and a rapid disease course. Patients who are simultaneously positive for anti-GM1 and anti-GD1a antibodies are relatively rare. Among 119 patients with GBS, one study identified only two patients who were simultaneously positive for immunoglobulin G (IgG) or immunoglobulin M (IgM) anti-GM1 and IgG anti-GD1a antibodies.2 In addition, overlap of AMAN and acute transverse myelitis (ATM) can have severe symptoms with poor prognosis.5 Although a recent study reported that patients with antibodies against a certain type of ganglioside complex experience severe GBS symptoms and outcomes,3 clinical manifestations and prognosis of patients with GBS overlapped with those of ATM who are simultaneous positive for both anti-GM1 and anti-GD1a antibodies remain poorly established.
Here, we report a clinical manifestation and prognosis of a 41-year-old male diagnosed with AMAN and ATM, positive for IgM anti-GM1, IgG anti-GM1, and IgG anti-GD1 antibodies. After rapid progression, he showed a favorable outcome after intravenous immunoglobulin (IVIG) and IV steroid therapy.
CASE
A previously healthy 41-year-old male presented with initial symptoms of bilateral leg weakness and paresthesia, which began 1 day prior to admission. He had experienced diarrhea and chills 1 week earlier. These was no autonomic dysfunction, including bladder and bowel function, body temperature, heart rate, or blood pressure. Laboratory findings revealed mildly increased creatinine (1.14 mg/dL), increased erythrocyte sedimentation rate (24 mm/hour), and normal C-reactive protein (0.5 mg/dL). Other laboratory findings such as lipid profile, electrolyte, liver function, and coagulation profiles were unremarkable. Cerebrospinal fluid analysis showed a white blood cell count of 5/mm3, glucose of 60 mg/dL, and protein of 25 mg/dL. Albuminocytologic dissociation was not seen.
Initial neurological examination revealed a medical research council (MRC) grade of -5 in the right upper limb, grade 5 in the left upper limb, grade 2 in the right lower limb, and grade 3 in the left lower limb. He showed paresthesia of both limbs, gait disturbance, and areflexia. The Romberg sign was negative and no ataxic symptoms were noted. Under suspicion of GBS, intravenous immunoglobulin was started immediately. Within 2 days, his symptoms rapidly progressed to MRC grade 3 in both proximal upper limbs and showed bilateral hand grip weakness and MRC grade 2 in both lower limbs. Laboratory tests confirmed the presence of anti-GM1 IgG, anti-GM1 IgM, and anti-GD1a IgM antibodies. Immunologic laboratory findings were negative for serum immunoglobulin A (287.4 mg/dL), anti-cardiolipin antibodies, aquaporin-4 antibodies, and anti-β2-glycoprotein I antibodies, while the lupus anticoagulant screening test was positive. Campylobacter species were detected in the stool.
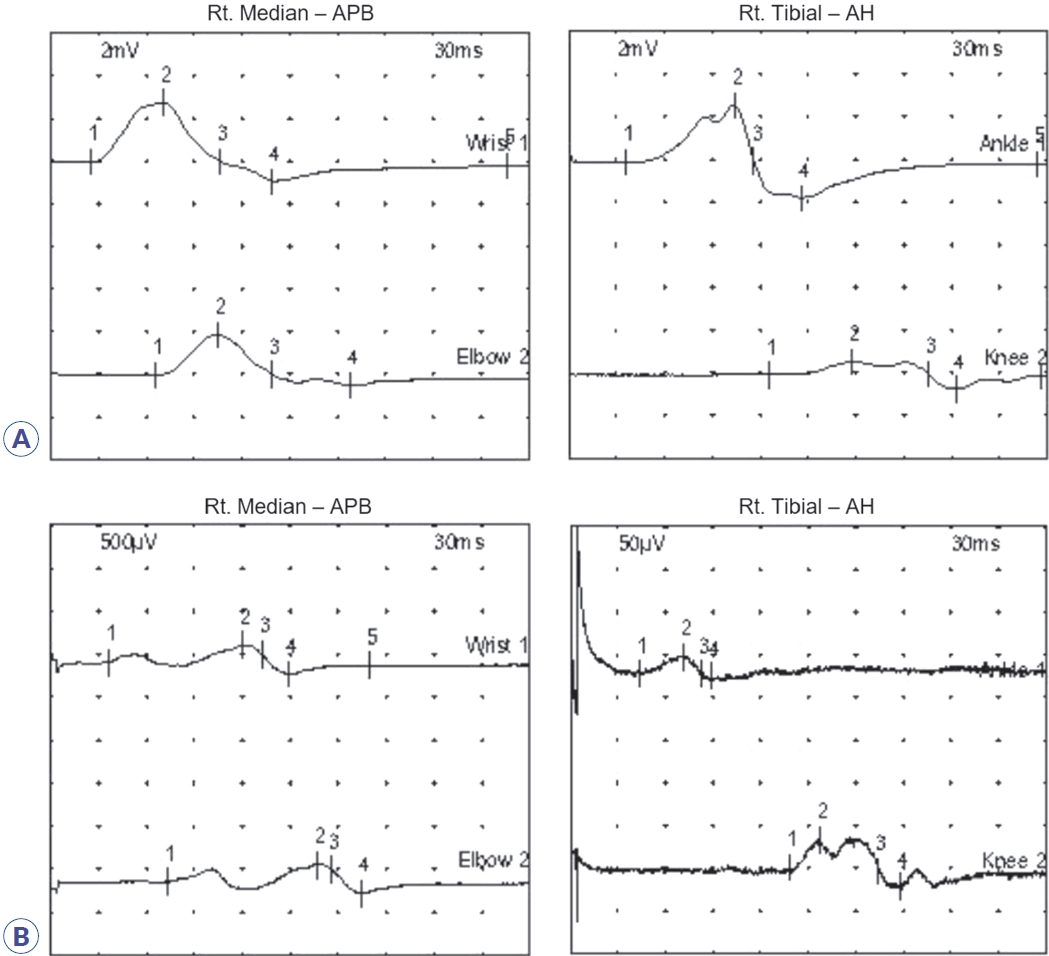
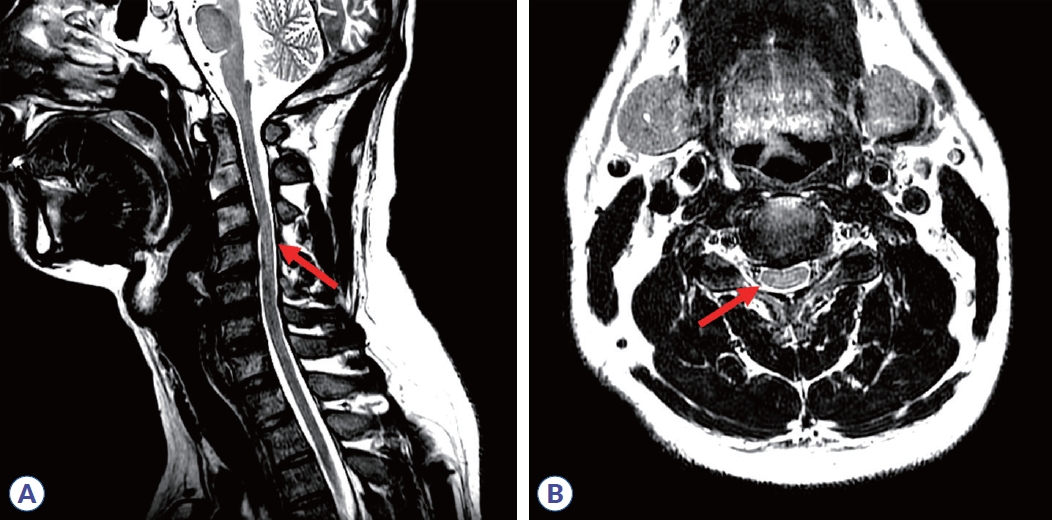
Initial nerve conduction studies supported polyneuropathy of motor nerves (Table 1, Fig. 1A), though it was unclear whether it was of demyelinating or axonal type. Conduction block and temporal dispersion were noted, indicating an early stage of demyelination of motor nerves. Further decrement of the amplitude of compound muscle action potentials without temporal dispersion or conduction block was noted after 5 days (Table 2, Fig. 1B), suggesting axonal motor polyneuropathy. Despite 5 days of IVIG treatment, his symptoms continued to worsen. Due to suspected transverse myelitis observed on cervical spinal magnetic resonance imaging (Fig. 2), steroid pulse therapy (methylprednisolone, 1,000 mg/day for 5 days) was initiated. An aquaporin-4 autoantibody test was negative. After initiating steroid therapy, no further progression was noted. Concurrent azithromycin treatment was added to combat the Campylobacter infection.
At 2 weeks after admission, the patient was discharged with a prescription for prednisolone. At discharge, neurological examination revealed MRC grade 4 in both proximal upper extremities, grade 3 in both distal upper extremities, and grade 2 in both lower extremities. His symptoms gradually improved over time. At 6 months, neurological examination revealed MRC grade 5 in both upper extremities (only some clumsiness) and grade 3 in the right lower and grade 4+ in the left lower extremities, allowing the patient to perform most daily activities, including walking.
DISCUSSION
Here, we present a patient diagnosed with the AMAN subtype of GBS who was simultaneously positive for both anti-GM1 and anti-GD1a antibodies and presented co-morbid acute transverse myelitis in the cervical spine. Anti-GM1 and anti-GD1 antibodies cause peripheral axonal polyneuropathy.6 Anti-ganglioside antibody on the basis of molecular mimicry theory make us understand association between types of autoantibody and patient’s clinical symptoms. Associated transient conduction block can be reversed in many cases of AMAN type GBS.6
Acute inflammatory demyelinating polyneuropathy is a demyelinating subtype of GBS, though the specific pathogens involved and their effects on myelin or Schwann cells are not clear.6 Thus, it is likely that the presented case of acute transverse myelitis in the cervical spine occurred through an unknown autoimmunologic mechanism. Transverse myelitis is a clinical syndrome that involves damage to the spinal cord and nerves resulting in various degrees of weakness, sensory alterations, and autonomic dysfunction that can be triggered by post-infectious immune system mechanisms similar to those of GBS.7 As such, corticosteroid therapy can be used to overcome the underlying inflammatory process. The presented patient experienced rapid progression of motor weakness to nearly quadriplegia within a few days despite IV immunoglobulin treatment but showed more favorable outcomes after initiation of steroid therapy. Substantial improvement was observed over the 6 months of steroid therapy. This case emphasizes the importance of recognizing the diverse clinical presentations and antibody profiles in GBS to allow prompt and appropriate therapeutic interventions. The presence of specific anti-ganglioside antibodies in GBS, particularly in the AMAN subtype, might contribute to the associated axonal dysfunction. In addition, simultaneous positivity for both anti-GM1 and anti-GD1a antibodies may play a role in rapid disease progression. As exact titers of single or complex antibodies were not analyzed, a causal relationship could not be established for the current case. It is also possible that ATM is the reason for the initial severe symptoms and the effectiveness of steroid therapy.5
The improvements observed during the 6 months of IVIG and steroid therapy, in conjunction with targeted treatment for an identified Campylobacter infection, underscore the importance of a multifaceted approach for patients with GBS. Although Campylobacter infections are usually self-limiting, in cases of invasive illness, and antibiotics such as azathioprine can shorten the duration of symptoms.8 In the presented case, evaluation of myelin oligodendrocyte glycoprotein antibody or follow-up anti-ganglioside antibodies might have allowed more stronger conclusion regarding the prognosis.
This case of AMAN-type GBS with both anti-GM1 and anti-GD1a antibodies highlights the interplay among infectious triggers, immune responses, and neurological manifestations. Despite early intervention with appropriate treatment, the disease and associated weakness can deteriorate rapidly. Steroid treatment was effective in the presented case of AMAN-type GBS with co-morbid ATM due to the involved axonal loss.5
This case demonstrated the possibility of substantial improvements over several months of such treatment, although the weakness had progressed to near quadriplegia. Accordingly, comprehensive diagnostic evaluations are crucial to differentiate among GBS subtypes and other demyelinating diseases to achieve optimal patient care and outcomes.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print